
An open-source pipeline to reconstruct phylogenies with paleoproteomic data
 based on reviews by Katerina Douka and 2 anonymous reviewers
based on reviews by Katerina Douka and 2 anonymous reviewers
PaleoProPhyler: a reproducible pipeline for phylogenetic inference using ancient proteins
Abstract
Recommendation: posted 01 September 2023, validated 19 September 2023
Hlusko, L. (2023) An open-source pipeline to reconstruct phylogenies with paleoproteomic data. Peer Community in Paleontology, 100220. https://doi.org/10.24072/pci.paleo.100220
Recommendation
One of the most recent technological advances in paleontology enables the characterization of ancient proteins, a new discipline known as palaeoproteomics (Ostrom et al., 2000; Warinner et al., 2022). Palaeoproteomics has superficial similarities with ancient DNA, as both work with ancient molecules, however the former focuses on peptides and the latter on nucleotides. While the study of ancient DNA is more established (e.g., Shapiro et al., 2019), palaeoproteomics is experiencing a rapid diversification of application, from deep time paleontology (e.g., Schroeter et al., 2022) to taxonomic identification of bone fragments (e.g., Douka et al., 2019), and determining genetic sex of ancient individuals (e.g., Lugli et al., 2022). However, as Patramanis et al. (2023) note in this manuscript, tools for analyzing protein sequence data are still in the informal stage, making the application of this methodology a challenge for many new-comers to the discipline, especially those with little bioinformatics expertise.
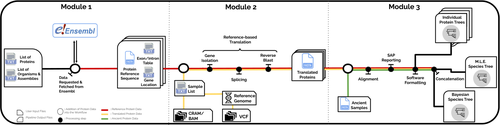
In the spirit of democratizing the field of palaeoproteomics, Patramanis et al. (2023) developed an open-source pipeline, PaleoProPhyler released under a CC-BY license (https://github.com/johnpatramanis/Proteomic_Pipeline). Here, Patramanis et al. (2023) introduce their workflow designed to facilitate the phylogenetic analysis of ancient proteins. This pipeline is built on the methods from earlier studies probing the phylogenetic relationships of an extinct genus of rhinoceros Stephanorhinus (Cappellini et al., 2019), the large extinct ape Gigantopithecus (Welker et al., 2019), and Homo antecessor (Welker et al., 2020). PaleoProPhyler has three interacting modules that initialize, construct, and analyze an input dataset. The authors provide a demonstration of application, presenting a molecular hominid phyloproteomic tree.
In order to run some of the analyses within the pipeline, the authors also generated the Hominid Palaeoproteomic Reference Dataset which includes 10,058 protein sequences per individual translated from publicly available whole genomes of extant hominids (orangutans, gorillas, chimpanzees, and humans) as well as some ancient genomes of Neanderthals and Denisovans. This valuable research resource is also publicly available, on Zenodo (Patramanis et al., 2022).
Three reviewers reported positively about the development of this program, noting its importance in advancing the application of palaeoproteomics more broadly in paleontology.
References
Cappellini, E., Welker, F., Pandolfi, L., Ramos-Madrigal, J., Samodova, D., Rüther, P. L., Fotakis, A. K., Lyon, D., Moreno-Mayar, J. V., Bukhsianidze, M., Rakownikow Jersie-Christensen, R., Mackie, M., Ginolhac, A., Ferring, R., Tappen, M., Palkopoulou, E., Dickinson, M. R., Stafford, T. W., Chan, Y. L., … Willerslev, E. (2019). Early Pleistocene enamel proteome from Dmanisi resolves Stephanorhinus phylogeny. Nature, 574(7776), 103–107. https://doi.org/10.1038/s41586-019-1555-y
Douka, K., Brown, S., Higham, T., Pääbo, S., Derevianko, A., and Shunkov, M. (2019). FINDER project: Collagen fingerprinting (ZooMS) for the identification of new human fossils. Antiquity, 93(367), e1. https://doi.org/10.15184/aqy.2019.3
Lugli, F., Nava, A., Sorrentino, R., Vazzana, A., Bortolini, E., Oxilia, G., Silvestrini, S., Nannini, N., Bondioli, L., Fewlass, H., Talamo, S., Bard, E., Mancini, L., Müller, W., Romandini, M., and Benazzi, S. (2022). Tracing the mobility of a Late Epigravettian (~ 13 ka) male infant from Grotte di Pradis (Northeastern Italian Prealps) at high-temporal resolution. Scientific Reports, 12(1), 8104. https://doi.org/10.1038/s41598-022-12193-6
Ostrom, P. H., Schall, M., Gandhi, H., Shen, T.-L., Hauschka, P. V., Strahler, J. R., and Gage, D. A. (2000). New strategies for characterizing ancient proteins using matrix-assisted laser desorption ionization mass spectrometry. Geochimica et Cosmochimica Acta, 64(6), 1043–1050. https://doi.org/10.1016/S0016-7037(99)00381-6
Patramanis, I., Ramos-Madrigal, J., Cappellini, E., and Racimo, F. (2022). Hominid Palaeoproteomic Reference Dataset (1.0.1) [dataset]. Zenodo. https://doi.org/10.5281/ZENODO.7333226
Patramanis, I., Ramos-Madrigal, J., Cappellini, E., and Racimo, F. (2023). PaleoProPhyler: A reproducible pipeline for phylogenetic inference using ancient proteins. BioRxiv, 519721, ver. 3 peer-reviewed by PCI Paleo. https://doi.org/10.1101/2022.12.12.519721
Schroeter, E. R., Cleland, T. P., and Schweitzer, M. H. (2022). Deep Time Paleoproteomics: Looking Forward. Journal of Proteome Research, 21(1), 9–19. https://doi.org/10.1021/acs.jproteome.1c00755
Shapiro, B., Barlow, A., Heintzman, P. D., Hofreiter, M., Paijmans, J. L. A., and Soares, A. E. R. (Eds.). (2019). Ancient DNA: Methods and Protocols (2nd ed., Vol. 1963). Humana, New York. https://doi.org/10.1007/978-1-4939-9176-1
Warinner, C., Korzow Richter, K., and Collins, M. J. (2022). Paleoproteomics. Chemical Reviews, 122(16), 13401–13446. https://doi.org/10.1021/acs.chemrev.1c00703
Welker, F., Ramos-Madrigal, J., Gutenbrunner, P., Mackie, M., Tiwary, S., Rakownikow Jersie-Christensen, R., Chiva, C., Dickinson, M. R., Kuhlwilm, M., De Manuel, M., Gelabert, P., Martinón-Torres, M., Margvelashvili, A., Arsuaga, J. L., Carbonell, E., Marques-Bonet, T., Penkman, K., Sabidó, E., Cox, J., … Cappellini, E. (2020). The dental proteome of Homo antecessor. Nature, 580(7802), 235–238. https://doi.org/10.1038/s41586-020-2153-8
Welker, F., Ramos-Madrigal, J., Kuhlwilm, M., Liao, W., Gutenbrunner, P., De Manuel, M., Samodova, D., Mackie, M., Allentoft, M. E., Bacon, A.-M., Collins, M. J., Cox, J., Lalueza-Fox, C., Olsen, J. V., Demeter, F., Wang, W., Marques-Bonet, T., and Cappellini, E. (2019). Enamel proteome shows that Gigantopithecus was an early diverging pongine. Nature, 576(7786), 262–265. https://doi.org/10.1038/s41586-019-1728-8

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
The project was funded by the European Union’s EU Framework Programme for Research and Innovation Horizon 2020, under Grant Agreement No. 861389- PUSHH. FR was additionally supported by a Villum Young Investigator Grant (project no. 00025300), a COREX ERC Synergy grant (ID 951385) and a Novo Nordisk Fonden Data Science Ascending Investigator Award (NNF22OC0076816). E.C. was additionally supported by the European Research Council (ERC) through the ERC Advanced Grant ”BACKWARD”, under the Eu- ropean Union’s Horizon 2020 research and innovation program (grant agreement No. 101021361)
Evaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2022.12.12.519721
Version of the preprint: 1
Author's Reply, 25 Aug 2023
Decision by Leslea Hlusko, posted 17 Jul 2023, validated 17 Jul 2023
Thank you for your patience as we located reviewers for your manuscript and gave them time to read and implement the pipeline. We now have three reviews (2 anonymous and 1 signed) that are presented in the spirit of advancing science respectfully and thoughtfully. All three are very supportive of your development and public posting of PaleoProPhyler. While one reviewer ran into difficulting executing two of the three modules in the pipeline, this reviewer was encouraging of your approach. All three reviewers offer specific advice on how to improve your manuscript, including a more detailed description of the three modules. As you prepare your revision, please include a response to the reviewers. I look forward to reading your revision.
Reviewed by anonymous reviewer 1, 24 Mar 2023
Patramanis et al. describe PaleoProPhyler, a pipeline to download, build, and analyze protein sequence databases for phylogenetics including with paleoproteomic sequences. This is an interesting workflow and will help standardize phylogenetics in paleoproteomics.
“files into amino acid seuqences” should be “files into amino acid sequences”
Description of the Pipeline: Please add detail/summary of each module here in the main manuscript. The supplementary has nice detail of each module, but it is very lacking here in the main manuscript.
Supplementary Choosing and preparing the list of proteins: \Reference_Protein_List.txt is not present in the github repository
Supplementary Final Execution: This section feels incomplete. I’m not sure what is needed, but more detail is probably helpful.
Reviewed by Katerina Douka, 20 Apr 2023
This is an exciting development in the field of palaeoproteomics and one that the community will welcome. I recomment the manuscript for publication and include below my comments and some minor corrections/additions.
-------
1/ To maket the manuscript appear more informed, I would add in the first paragraph that while shotgun proteomics is used to infer phylogenic relationships, another palaeoproteomics method (PMF or ZooMS when for collagen) is used as a primary tool for identifying new hominid remains, which can then be analysed deeper with shotgun proteomics, ultimately using the new bioinformatics tool presented here.
If so, I would add a few more references aside from the Copenhagen group. We are talking about democratisation of the field, citing more widely is part of it too. I believe the oldest collagen analysed so far is presented in Rybczynski et al. (2013) also more recently expanded (Buckley et al. 2020, cited already), and other teams have also published very ancient proteins (e.g Nielsen-Marsh et al. 2009 (https://www.sciencedirect.com/science/article/pii/S0305440309001253), or Brown et al. 2022 (https://www.nature.com/articles/s41559-021-01581-2).
2/ Page 2. "The amount of publicly available proteome sequences is much smaller in comparison".-> Can you quantify this? There are indeed very few.
3/ For Module 3, I would have appreciated comments on thresholds or limitations for the use of PaleoProPhyler. Are there are any? What are the limitations imposed by the (often) small number and of poor preservation of proteins/peptides for a given sample. Are there cut-offs and suggestions how to overcome them?
4/ There is a mention for Supplementray Material, I could not see it or access it.
5/ Unless there is a very specific word limitation, there is very little in the description of how the pipeline works and even what each Module does. I like the graphical abstract but I was left wondering where is the input and output and, as alaready mentioned, indication of cut-offs and generaly data hygene.
Some minor stuff:
“…lab-generated protein data does not even exist” : Remove even
“…absence of knowledge about even a single amino acid polymorphism”: Remove even
“The modules are intended to synergize with each other” : I am not sure of the word synergize here. Maybe best to keep it simple and say “work with each other”
https://doi.org/10.24072/pci.paleo.100220.rev12
Reviewed by anonymous reviewer 2, 17 Jul 2023
The manuscript ‘PaleoProPhyler: a reproducible pipeline for phylogenetic inference using ancient proteins’ by Patramanis and colleagues presents an open-source pipeline for the phylogenetic analysis of palaeoproteomic data. The pipeline is split into three modules which follow on from each other, but can be run independently. These build a basic reference database from proteomes available on Ensembl (module 1), transcribe published genomes to supplement the reference database (module 2), and perform phylogenetic analysis of proteomic data using the reference database (module 3). The motivation for the development of the pipeline and a brief overview are provided in the main text with a more detailed explanation of the workflow presented in the supplementary information and the code available on the github of the lead author. A tutorial is provided to train users in how to install and run the pipeline using published data to reconstruct the enamel phylogeny of two hominids, Homo antecessor (Welker et al 2020) and Gigantopithecus blacki (Welker et al 2019). The authors used modules 1 and 2 of the pipeline to curate a hominid palaeoproteomics reference database which they make publicly available on Zenodo.
Open-source tools for reproducible data processing and analysis between different research groups and labs are important areas of development for the field of palaeoproteomics, as they are currently lacking. This hinders data reproducibility and represents a barrier to researchers within the field who lack formal training in computational biology. The PaleoProPhyler pipeline presented by the authors addresses this issue and therefore has the potential to be a timely and important addition to the toolset available to the palaeoproteomics community. The rationale for the work is clear and the manuscript is well written. The modularity of the pipeline is highly useful and will enable users to adopt portions of the pipeline for their own uses. The tutorial written with a ‘non-bioinformatics-background audience in mind’ is an excellent resource to increase accessibility to a wide range of researchers and achieve the aim of improving reproducibility within the field.
I am not a bioinformatician so will not comment on the scripts themselves but will comment from the point of view of the ‘non-bioinformatics’ audience, the target audience of the tutorial. Unfortunately, I was only able to run the first module of the pipeline when following the tutorial, whilst Modules 2 and 3 resulted in errors and termination of the script. Perhaps readers with bioinformatics training would be able to adapt the scripts to make them run but even with access to server and bioinformatics support I was unable to complete the tutorial. Therefore, to be widely employed by researchers with different computational setups, some revisions to minimise dependencies and the potential for clashes between systems would be beneficial.
The tutorial first directs the user to download the github workflow and install the conda environment from the command line and then download the published fasta files of the two hominid proteomes. As noted by the authors, the user ideally needs access to a high performance lab server for sufficient computational power to run the pipeline. The installation of the conda environments and pipeline may clash with pre-installed software on the institutional server which the user has no access to modify. This may act as a barrier to the installation of the pipeline.
The first module generates a scaffold reference database by downloading proteomes from species closely related to the hominids from Ensembl, a publicly available database for annotated genomic data. The second module is designed to supplement the scaffold reference database through the transcription of published genomic data, including other ancient hominins.
Running the first module was relatively quick and straightforward. Some further information or references could be added on the strengths/weaknesses of downloading reference proteomes from Ensembl vs translating genomes. I was unable to run the second module so cannot comment on the output.
The third module merges together the palaeoproteomic data with the reference datasets and performs phylogenetic analysis. Implementing the module seems very straight-forward, however the tutorial ends abruptly after the analysis has been run with no further information on where the output files are generated. The tutorial could be improved by adding additional information here on how to check the output of the analysis (as the authors did at the end of module 1), how to visualise the trees generated data and some simple QC checks to carry out.
Although the pipeline may run successfully on the author’s institutional server, it needs to be packaged more efficiently for widespread use. There appears to be some typos in the code or system incompatibility which prevent the pipeline from running to completion. It would require a bioinformatician to troubleshoot the errors. This is therefore a barrier to anyone without this knowledge base.
This is a common problem when sending code between labs and can require some complicated trial and error to solve. I suggest packaging the software into a container so it can be shared between labs without issues of installation in clashing systems. Enlisting several researchers from different labs outside the Globe Institute to install and run the pipeline tutorial on their own servers would provide the authors with the opportunity to trouble-shoot any issues that arise.
Other points:
- The system requirements for running the pipeline on a linux OS are not apparent until the SI and tutorial - this could be mentioned in the main text under ‘Availability and Community Guidelines’.
- The hominid reference database will be highly useful. Although the references for the data are available in the SI, a table with all of the individuals included would be useful.
- Overall the authors have done a good job adding useful tips, warnings, additional descriptions and links to resources to help users who are new to this type of analysis. Perhaps a text box with a glossary/key terms to provide additional descriptions of the different file types (FASTA, VCF, BAM. CRAM) could be useful for a non-bioinformatics audience, as there are lots of abbreviations used.
- Ref 61 in the first paragraph of the Statement of Need appears to have no link.
- There are some typos throughout the tutorial text so some proofreading would be beneficial.




